![]()
Disrupted brains without autism--important but overlooked clue to the cause of autism
The thalidomide-autism anomaly--overlooked etiological clue
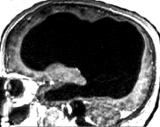 non-autistic brain |
This picture is not
of an individual with autism, but it is key for learning the secret
of autism.
The brain scan is of a married father of two children, who works as a
civil servant in France. Apart from slight problems in moving his left
leg when young, his "neurological development and medical history were
otherwise
normal". The brain scan shows that he has "massive enlargement of the
lateral, third, and fourth ventricles, a very thin cortical mantle" due
to hydrocephalus. Here for a pdf. The existence of this man
shreds light on autism in two respects. |
| First,
people can have grossly abnormal brains that have little white
matter and very thin cortical mantle but not autism. Second, autism researcher do not know the neuroscience that they should. If they were curiosity driven scientists, they would have known about this individual (and other similar cases), and not proposed a neurobiology that attributed its cause to cerebral connection defects. |
|
 |
This happened in 2007 in the Archives of Neurology.
It published a review titled "The new neurobiology of autism: cortex,
connectivity, and neuronal organization". This claimed what should
never have got into print: that autism links "to
a disorder of the association cortex, both its neurons and their
projections. In particular, it is a disorder of connectivity
...
to primarily .. intrahemispheric
connectivity. The focus of connectivity studies thus far has been on
white matter, but ... intracortical connectivity is also likely to be
disturbed." Read the abstract or pdf. It should not because that
French civil servant directly refutes that idea as I pointed out later
in the Archives of
Neurology here.
|
 |
Remarkably, there is no neurological
defective that links specifically to autism. Many associations exist
but in all
cases individuals without autism can be found with them as well.
No report exists of a neurological factor that only occurs in
those with autism. This situation is hidden by the use of group
comparisons that statistically down play that the fact many
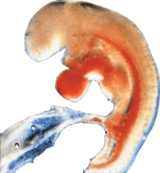
individuals without autism exist with the factor attributed to autism.The nearest specific brain autism link is that autism can occurs in those exposed to
thalidomide at 20-24
days gestation, a time at which most of the brain has yet to form. |
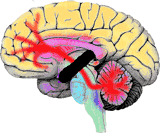 |
This
identifies its cause to disruption in the links between the cerebral
cortex and
the cerebellum. |
The teratogen thalidomide induces autism at 20-24 gestation days (10). At this date the embryo is roughly the size of this `C', and the brain (apart from the neurons of the cranial motor never nuclei) does not yet exist (11). Indeed, at this stage, the neural tube, out of which the future brain will arise, is only just closing. This dating of the thalidomide-autism link is precise: thalidomide is teratogenic roughly between gestation days 20 and 35 (12) with various impairments linking to specific gestation days. Between days 20-22, for instance, the outer ear is affected, between 22 and 28, the thumb fails to develop, the upper limbs are affected between days 24 and 31, the lower ones on days 27 to 33, and between days 32 and 36, the thumb has an extra joint (10). These and other stigmata precisely date the period during which thalidomide causes autism. Many factors later in embryo or even postnatal development might cause the neuropathology responsible for autism. But whatever neuropathology they induce, it logically must be one that can also be induced by impairments at this early stage.
Why anomalous
Many functions impaired in autism depend upon higher brain areas that only arise after the early gestation date of thalidomide-induced autism. For example, those of the human cerebral cortex arise at the end of the fifth week, for the hippocampus at the beginning of the seventh week, and for the amygdala and the hypothalamus, they arise at the middle of the fifth week, even the neurons for the cerebellum and its nuclei only first arise at the middle of week five (13). Effects upon stem cells generating them are unlikely since such impairment would lead to acephalia (stem cells also form the bone and other tissues that later surround the brain) (14), something which does not happen. Autistic brains, if anything, tend to be larger than normal (15). This makes it anomalous how impairments at 20-24 days could have specific neurological consequences upon functions that are impaired in autistic individuals such as theory of mind that take place in the prefrontal cortex.
The autism cerebellar disconnection hypothesis
Many higher cognitions depend upon the communication between the cerebral cortex and the cerebellum, and that, due to the peculiarities of their development, these connections are vulnerable to brainstem disruption. Disruption to the cerebral-cerebellar link can arise at many developmental stages - not only at the early date proposed for thalidomide (this, though, is theoretically interesting, because it identifies this connection as the target neuropathology underlying autism).
Autism is due to disconnection of cerebral input (including the prefrontal cortex) to the cerebellum. Skills in the prefrontal cortex depend upon an active interaction with processes in the cerebellum (6). Skills such as theory of mind, that are selectively impaired in autism, I suggest, are those that are particularly dependent upon interactions between these two areas of the brain. Other skills preserved in autism are not affected because they can function in the absence of cerebral-cerebellum interaction.
Axons from the cerebral cortex go to the cerebellum via a projection to neurons in the pons. However, due to peculiarities in their development, the axons going down to the pons, initially go down further to the lower medulla and the cranial nuclei in the brainstem.
It is only after growing down into the brainstem, that axon collaterals bud off and enter the pons. (Many of the initial brainstem axons indeed survive - thus the somatomotor cortex projection to the cerebellum is bifurcate and projects both to the pons and spinal and brainstem motorneurons.) However, the initial axons from other cerebral cortex areas (such as the visual cortex) die off leaving only the pons projection (16). Interestingly, a transient link from the cerebral cortex to the cerebellum exists that goes direct to it without first going to the pons: it follows the same developmental pattern of developing as a collateral to an initial brainstem projection (17). This suggests that the collateral strategy of development for reasons unknown is a developmental rule for axons developing into the pons/cerebellar region from the cerebral cortex. Due to this initial projection to the brainstem, impairments to the brainstem could knock-on and affect the cerebral cortex-cerebellum link: this vulnerability arises because excess cerebral-pons axons (depending upon their integrity) are eliminated through `axon sculpturing'. This elimination (at least in the rat), for example, proceeds first in a rapid period during one week in which over a third are removed, and second, in a slower period that continues until only half the original axons remain (18). Two factors determine the integrity of axons, and hence their survival, that link to earlier brainstem events. First, axons reach targets both cells (19) and intermediate substrates (20) that provide retrograde transported trophic factors (such as NGF). Second, axon connections with neurons from an early date are active (neurons in the rat brainstem, for example, are active in `respiratory oscillations' from gestation day E 18) (21). Such electrical activity is known to determine cerebral-brainstem and cerebellar related axon survival, for instance, lack of it is responsible for the direct cortical-cerebral link noted above being transient (22). These factors act to make axon sculpturing potentially sensitive to any earlier compromise of the brainstem. Thus, impaired connection by axons to neurons in the brainstem caused by defects to these neuron targets could knock-on and impair the integrity of the later developing cerebral cortex-cerebellum axon links.
Axon input from the frontal areas of the cerebral cortex would be particularly affected by this knock-on effect. Collaterals from the occipital and frontal regions of the cortex arrive after those from lateral areas. As Leergaard and colleagues note: `They grow around the central core [into which early arriving axons go], bridging [i.e. infilling] the rostral and caudal entrance zones' (23). A general process in neurological development is weeding out by competition (24). Thus competition for neuron targets in the pons in the sculpturing process will delete excess axons. This will put axons from occipital and frontal areas at risk from erroneous or excessive deletion: they arrive late, and are so likely to already be competing for pons neuron targets already taken by the earlier-arriving lateral area ones. This would act to amplify any compromise upon them created by earlier impairment to the brainstem.
Development of input nuclei
A further possibility exists: the neurons which form the pons and the inferior olive nuclei that provide the cerebellum with its afferent input are generated in the dorsally located rhombic neuroepithelium and migrate from this lower and early developed brainstem region up to their final upper brainstem location (25). Early effects of thalidomide might affect either the integrity of their neurogenesis or their receptor expression. Again here is an opportunity to affect the input from the cerebral cortex to the cerebellum. This is less likely since though research finds abnormalities in these neurons in regard to their size - they fail to find neuron number reduction in these nuclei in autistic brains (5). However, it is possible that subtle impairment in the brainstem might leave them apparently intact though of smaller size and compromised in their function abilities (though this is not the interpretation of the researchers).
Autism and the brainstem
Thalidomide impairs the development of axons from the cerebral cortex to the cerebellum because of the sensitivity and vulnerability of this link to brainstem nuclei compromise. Later in development, other impairments (of other causes) might also impair this link, so producing autism of other origins. Thalidomide, however, by causing impairment at 20-24 days gestation points the finger of suspicion to its critical neuropathological impairment being dysfunction in the link from the cerebral cortex to the cerebellum. Though, as noted above, this connection could be impaired at times after 20-24 days, many autistic individuals interestingly seem to suffer a development compromise at this very early embryo stage. Stigma such as ear deformities known to link to this stage are common in autistic individuals (26). Moreover, autistic individuals show a high prevalence of a variant in Hoxa1 (40% compared to 20%), a gene that is involved in the development of the lower brainstem (27). However, such brainstem impairment, cannot itself be the cause of autism. Not all autistic individuals show such stigma, and the higher functions impaired in autism are selective: for example, it affects the ability to perceive the processes underlying intentional but not mechanical events (it would be difficult to see how a brainstem impairment could produce this). Moreover, many (though only a minority) of individuals with autism show normal levels of IQ, and even relatively normal lives (for example, one is a noted animal behaviourist and autobiographist, Prof. Temple Grandin (28). This argues that the explanation must be some impairment that does not hinder brainstem function (which would have clinically obvious and important effects) but affects a component needed for certain higher cognitions processed elsewhere in the brain (for example, those dependent upon the cerebral-cerebellar link).
This theory is not without precedence: present theories of autism link it to dysfunction in the cerebellum (29), prefrontal cortex (30) or even both (31). What is novel about this theory is that it argues its neuropathology links not to these areas but the uninvestigated connections between them. This is a more probable possibility than neuron-related theories - since impairments in these have been already been examined and not found. Cerebral cortex-cerebellum disconnection is plausible on the further grounds that it is the only major remaining defect that could impair the higher brain function that has yet to be investigated. The reason is technical: researchers at present see axon fibres connecting the cerebral cortex and the pons in detail only with the use of axon-carried biotracers. The result of this is that research upon them has been limited so far to experimental animals. Any abnormalities in this tract in human brains have thus, with the use of present techniques, been terra incognita. Therefore, if autism arose, as suggested from abnormalities in the cerebral-cerebellar link, we would expect its neuropathology to have remained, as it presently is, undiscovered.
Cited references
5. Bauman M. Microscopic neuroanatomic abnormalities in autism. Pediatrics 1991; 87(suppl.): 791-796.
6. Andreasen N. C., O'Leary D. S., Cizadlo T., Arndt S., Rezai K., Ponto L. L. et al. Schizophrenia and cognitive dysmetria: a positron-emission tomography study of dysfunctional prefrontal-thalamic-cerebellar circuitry. Proc Natl Acad Sci USA 1996; 93: 9985-9990.
10. Stromland K., Nordin V., Miller M., Akerstrom B., Gillberg C. Autism in thalidomide embryopathy: A population study. Develop Med Child Neurol 1994; 36: 351-356.
11. Rodier P. M., Ingram J. L., Tisdale B., Nelson S., Romano J. Embryological origin for autism: developmental anomalies of the cranial nerve motor nuclei. J Comp Neurol 1996; 370: 247-261.
12. Miller M. T. Thalidomide embryopathy. Trans Am Ophthol Soc 1991; 89: 623-574.
13. Bayer S. A., Altman J., Russo R. J., Zhang X. Timetables of neurogenesis in the human brain based on patterns in the rat. Neurotoxiology 1993; 14: 83-144.
14. O'Rahilly R, Muller F. The Embryonic Human Brain. New York: Wiley-Liss; 1994: pp. 52, 62.
15. Piven J., Arndt S., Bailey J., Havercamp S., Andreasen N. C., Palmer P. An MRI study of brain size in autism. Am J Psychiatry 1995; 152: 1145-1149.
16. O'Leary D. D., Terashima T. Cortical axons branch to multiple subcortical targets by
interstitial axon budding: Implications for target recognition and `waiting periods'. Neuron
1988; 1: 901-910.
17. Panneton W. M., Tolbert D. L. The collateral origin of a transient cerebro-cerebellar pathway in kittens. A study using fluorescent double-labelling techniques. Develop Brain Res 1984; 14: 247-254.
18. Stanfield B. B. The development of the corticospinal projection. Prog Neurobiol 1992; 38: 169-202.
19. Ure D. R., Campenot R. B. Retrograde transport and steady-state distribution of 125I-nerve
growth factor in rat sympathetic neurons. J Neurosci 1997; 17: 1282-1290.
20. Wang H., Tessier-Lavigne M. En passant neurotrophic action of an intermediate axonal target
in the developing mammalian CNS. Nature 1999; 401: 765-769.
21. Champagnat J., Fortin G. Primordial respiratory-like rhythm generation in the vertebrate embryo. Trends Neurosci 1997; 20: 119-124.
22. Tolbert D. L. Absence of impulse activity in cortical neurons with transient projections to the cerebellum. Develop Brain Res 1989; 50: 241-249.
23. Leergaard T. B., Lakke E. A., Bjaalie J. G. Development of projections from the cerebral cortex to the pontine nuclei. A Dil and computer 3-D reconstruction study in the rat. Soc Neurosci Abstr 1994; 20: 22.
24. Gordon N. Apoptosis (programmed cell death) and other reasons for elimination of neurons and axons. Brain Dev 1995; 17: 73-77.
25. Rodriguez C. I., Dymecki S. M. Origin of the precerebellar system. Neuron 2000; 27: 475-486.
26. Rodier P. M., Bryson S. E., Welch J. P. Minor malformations and physical measurements in autism: data from Nova Scotia. Teratology 1997; 55: 319-325.
27. Ingram J. L., Stodgell C. J., Hyman S. L., Figlewicz D. A., Weitkamp L. R., Rodier P. M. Discovery of allelic variants of HOXA1 and HOXB1: genetic susceptibility to autism spectrum disorders. Teratology 2000; 62: 393-405.
28. Grandin T. Thinking in Pictures. New York: Vintage Books 1995.
29. Courchesne E. Brainstem, cerebellar and limbic neuroanatomical abnormalities in autism. Curr Opin Neurobiol 1997; 7: 269-278.
30. Hughes C., Russell J., Robbings T. W. Evidence for executive dysfunction in autism.
Neuropsychologia 1994; 32: 477-492.
31. Carper R. A., Courchesne E. Inverse correlation between frontal lobe and cerebellum sizes in children with autism. Brain 2000; 123: 836-844.
For full paper see Is autism due to cerebral-cerebellum disconnection?